





Scientists and clinicians expect that an understanding of the disease mechanisms and ensuing molecular defects can deliver novel therapeutic targets. Cardiac hypertrophy, being an independent clinical risk factor for cardiac death,
has been the subject of intensive research throughout the past 2 decades. Despite numerous advocated targets derived from molecular insight into maladaptive hypertrophic growth, no effective therapeutic strategy has been forthcoming to date. The reason may be that basic research does not provide us per se with a roadmap of how to translate our knowledge therapeutically. The complexity of the manipulated biological system, which is often incompletely understood, still poses the greatest challenge for rational design and preclinical development of new therapeutic modalities, such as gene-, cell-, or protein-based treatments.
Discovery of relevant disease mechanisms that impair cardiomyocyte performance, however, was more successfully used in designing gene-based inotropic therapies for HF. Although the pathogenesis of HF is diverse, common defects in β-AR signaling and Ca2+ cycling are key to HF pathogenesis. The therapeutic spectrum of inotropic HF gene therapies ranges from (1) direct substitution of a targeted protein by the gene product, as in the case of AC6, SERCA2a, or S100A1 to (2) the modulation of its function through an engineered oligopeptide, such as βARKct, being a G-protein–coupled receptor kinase 2 (GRK2) inhibitor, and (3) its indirect suppression by regulatory RNA intermediates as for phospholamban (PLN). Figure 7 illustrates underlying molecular mechanisms in failing cardiomyocytes. If corrected by cardiomyocyte-directed DNA/RNA delivery, the order of in vivo therapeutic effects is (1) regeneration of contractile performance of the failing heart and, because of improved peripheral perfusion, (2) subsequent reversal of neurohormonal overdrive and cardiac remodeling as demonstrated in numerous large animal HF models.
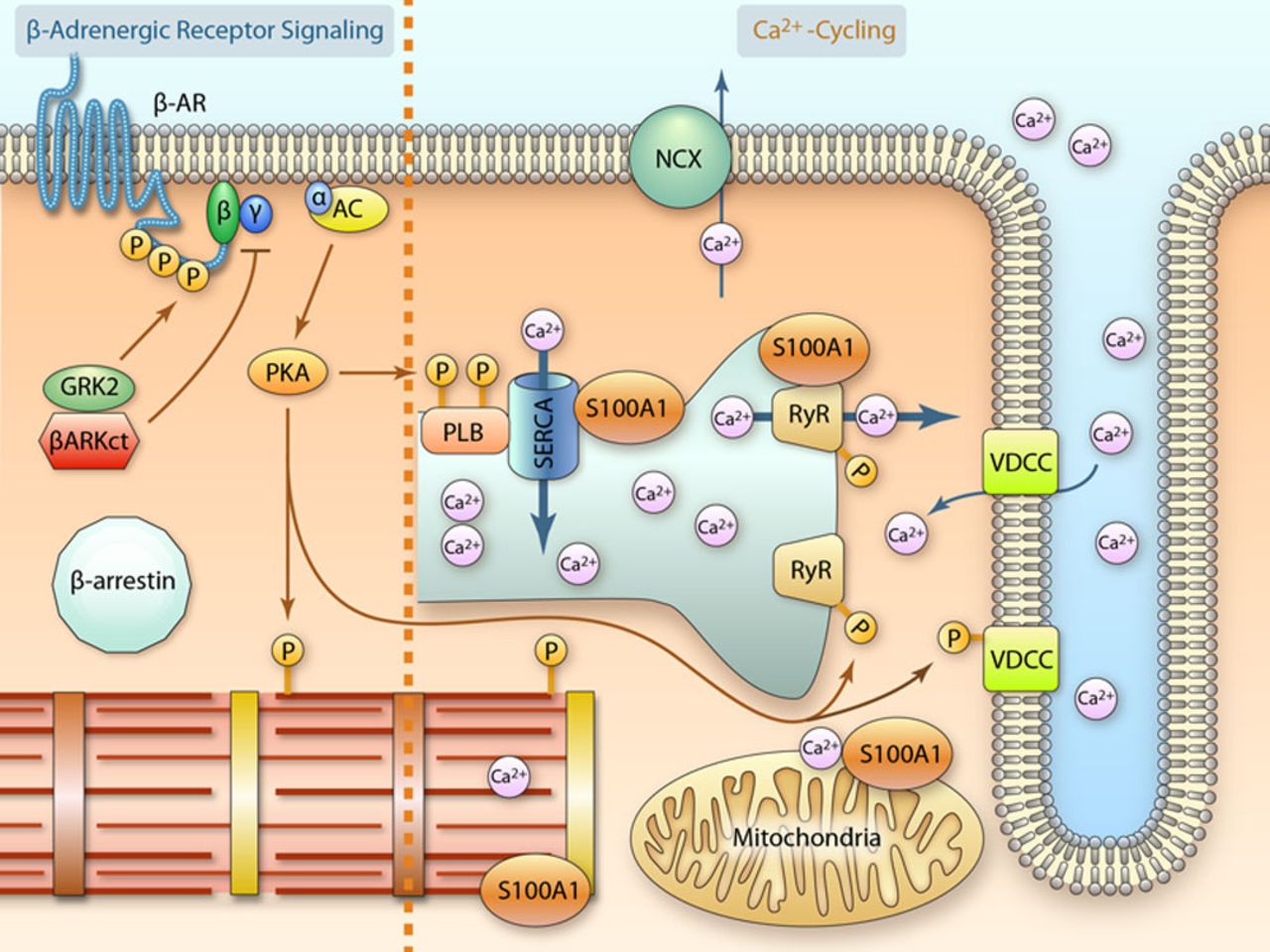
Figure 7.
Overview of selected inotropic targets for heart failure (HF) gene therapy. See DNA/RNA-Based HF Therapeutics Targeting Cardiac Inotropy section for detailed mechanistic information (illustration credit: Ben Smith). AC indicates adenylyl cyclase; β-AR, β-adrenergic receptor; βARKct, β-adrenergic receptor kinase C terminus; GRK2, G-protein–coupled receptor kinase 2; NCX, sodium/calcium exchanger (Na+/Ca2+ exchanger); PKA, protein kinase A; PLB, phospholamban; RyR2, ryanodine receptor 2; SERCA2a, sarco(endo)plasmic reticulum Ca2+ ATPase 2a; and VDCC, voltage-dependent calcium channel.
The central question eventually emerging from these concepts is, What makes a molecular HF target amenable to DNA/RNA-based treatment? A major prerequisite is that its abnormal function must be correctable through the chronic expression of the therapeutic DNA/RNA. But unlike pharmacological regimes where effective doses are freely adjustable, a yet unavoidable empiricism in the choice of vector and expression system results in unforeseeable and fixed levels of expression of the therapeutic DNA/RNA product. Vice versa, the therapeutic window of a DNA/RNA in the diseased heart is mostly unknown, unless gene-dosage studies are performed.
In other words, precise understanding of the DNA/RNA dose–response relationship seems mandatory to avoid premature dismissal of an otherwise effective gene product. Failure to test all emerging targets categorically and systematically is a major factor why the progress of HF gene therapy has been much slower than promised, and only a handful of the many reported potential targets have been developed toward clinically applicable treatments. An important lesson from the few successes is that only a refined translational pipeline sufficiently facilitates target-specific solutions that pave the way toward clinical translation (Figure 6). This section will integrate previously discussed data and focus on therapeutic inotropic targets with proven clinical applicability highlighting critical steps of their development.
β-Adrenergic System as Target for Gene Therapy
More than 20 years ago, diminished β1-AR density was demonstrated in explanted failing human hearts. Ensuing studies unveiled a loss of cAMP-dependent cardiac reserve mechanisms as a common defect in failing myocardium irrespective of the underlying cause because of β1-ARs internalization and desensitization. Clinical attempts aiming at improved cardiac contractility by administering chronic catecholamine or phosphodiesterase inhibitor therapy resulted in increased patient mortality; quite unexpected at this time. Now, comprehensive evidence suggests that pharmacological blockade of cardiac β-AR stimulation is actually life saving by shielding the vulnerable heart against noxious sympathoadrenergic overdrive.
Because βAR blockade has become a cornerstone of clinical practice, extensive research has been undertaken into the maladaptive molecular consequences of chronically activated β-ARs in the failing heart. Evidence gleaned from numerous experimental studies indicates that phosphorylation of the β1-AR cytoplasmic domain through GRK2, previously known as β-AR kinase 1 (βARK1), is a key event in β-AR desensitization. This leads to internalization of the receptor complex followed by attenuated Gs-protein coupling to AC, which entails diminished cAMP generation capacity. Although counterintuitive at first glance, inhibition of GRK2and improvement of AC activity surfaced as a potent strategy amenable to genetically targeted therapy (Figure 7) to restore β-AR–dependent cardiac performance without adverse effects.
Inhibition of GRK2
Failing human myocardium exhibits enhanced GRK2 expression and activity. A critical role of GRK2 in cardiac β-AR responsiveness was first shown by cardiac-targeted GRK2 overexpression in mice resulting in chronic β-AR desensitization. Cardiac-directed GRK2 deletion improved post-MI contractile performance basally and, as expected, in response to β-AR stimulation. Most importantly, post-MI outcome was improved by GRK2 ablation providing a clear rationale for the design of a GRK2 inhibitor, potentially suitable for gene-based therapy.
GRK2 requires Gβγ-dependent association with the cell membrane before β-AR phosphorylation. Because interaction with membrane-bound Gβγ subunits occurs via the C-terminal domain of the kinase, this prompted design of an engineered cDNA encoding the C terminus of GRK2, named βARKct, to compete with Gβγ binding and subsequently prevent β-AR phosphorylation (Figure 7). Proof-of-concept for augmented β-AR responsiveness was demonstrated in transgenic mice with cardiac-restricted βARKct expression. Ensuing functional rescue and improved survival of various genetic murine HF models by crossbreeding them with βARKct transgenic mice promoted βARKct as a therapeutic GRK2 inhibitor.
A DNA-based strategy was first tested in a rabbit model of post-MI HF with intracoronary delivery of an Ad harboring the CMV promoter–driven βARKct cDNA (AdβARKct) and it resulted in significantly improved β-AR responsiveness as well as AC-mediated cAMP production after 2 weeks. Subsequent experiments using AAV6 for cardiac-restricted βARKct gene therapy in an ischemic rat HF model demonstrated long-term therapeutic efficacy that was additive to pharmacological β-blocker treatment. A total of 2.5×1011 infectious particles of AAV6-βARKct were delivered via coronary arteries preceded by intracoronary application of a NO donor to facilitate vascular permeability and myocardial transduction. Achieving ca. 40% to 50% myocardial transduction rate, AAV6-βARKct not only restored cardiac β-AR density and signaling after 8 weeks but curtailed sympathetic overdrive, most likely because of sustained enhancement of myocardial performance. Equivalent to β-blocker treatment, AAV6-βARKct therapy exerted robust antihypertrophic actions in injured hearts supporting the notion that recovery of cardiac function is sufficient to reverse cardiac remodeling. Cardiomyocyte-targeted βARKct expression was assured by the use of a biased engineered expression cassette combining the cardiac actin enhancer with the elongation factor 1 promoter (Card-EF1).
The relevance of these studies was supported by improved β-AR responsiveness in AdβARKct-treated isolated human failing cardiomyocytes that were derived from patients with end-stage HF undergoing cardiac transplantation. Mechanistic studies detailed that βARKct’s immediate inotropic actions do not rely on enhanced cAMP production in normal or failing cardiomyocytes. It exerts its beneficial effects early on through a Gβγ-dependent but cAMP-independent enhancement of cardiomyocyte Ca2+ handling. As the ultimate step toward clinical translation, the long-term therapeutic impact and feasibility of AAV6-βARKct gene therapy was assessed in a human-relevant porcine model of ischemic cardiomyopathy. Cardiac-directed delivery of 1.5×1013 AAV6-βARKct particles 2 weeks after MI via the anterior coronary vein resulted in βARKct expression in the LV anterior wall, septum, and regions adjacent to the infarcted area. Chronic βARKct expression led to significant enhancement both of systolic and diastolic LV performance, reversed myocardial remodeling, and normalized catecholamine plasma levels after a 2-month follow-up. The use of an engineered cardiac-biased expression cassette coupling the CMV mini-enhancer to the MLC ventricle 2 promoter fragment (CMV-MLCV2) enabled cardiac-restricted expression of βARKct that was without negative impact or toxicity on peripheral organ functions and the hematopoietic system.
This systematic evaluation of βARKct’s therapeutic potency along a refined translational sequence provided striking evidence for its efficacy and safety in human-relevant HF models (Table). Because of a lack of quantitative data, an important question still remains whether the therapeutic effects seen in different HF models using various vectors and expression cassettes actually result from similar expression levels. Future studies evaluating the therapeutic profile of the Gβγ-sequester and GRK2 inhibitor should establish standards to quantify absolute βARKct expression levels in myocardium. Defining βARKct/GRK2 or βARKct/GAPDH ratios might help to determine effective gene dosages and lay the groundwork for an understanding of βARKct’s PK and PD. With growing understanding of its molecular actions that might actually extend far beyond GRK2 inhibition, βARKct-DNA–based therapy seems ready to enter phase I/II trials to evaluate its therapeutic potential in human HF.
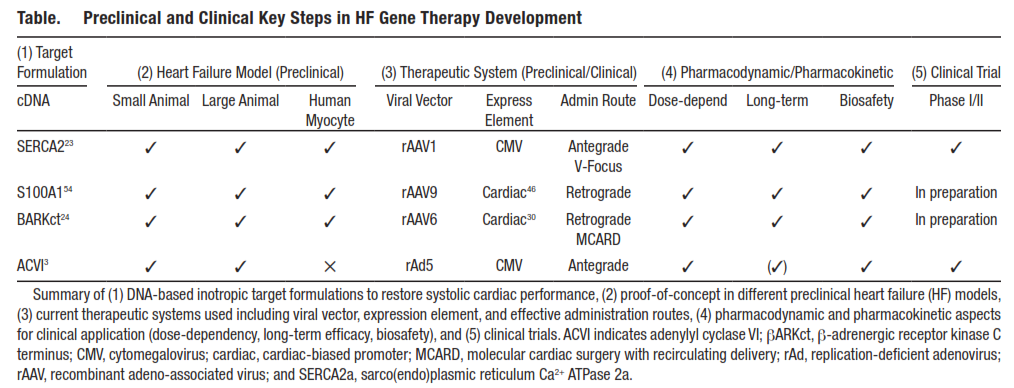
Table.
Preclinical and Clinical Key Steps in HF Gene Therapy Development
Improving AC6 Activity
As previously alluded, cAMP signaling plays a pivotal role in cardiac contractility. Pharmacological attempts to increase cAMP signaling in failing myocardium rendered patient hearts more susceptible to lethal tachyarrhythmias and increased mortality. Similarly, cardiac-directed expression of β1-AR, Gαs and protein kinase A, presumably because of sustained cAMP elevations, is deleterious and associated with cardiomyopathy in mice. It, therefore, surprises at first glance that expression of AC6, the enzyme catalyzing cAMP formation, exerts beneficial effects on the failing heart (Figure 7), but AC6 protein elevations do not effect basal cAMP production, and, additionally, may increase cardiac performance through cAMP-independent mechanisms.
Proof-of-concept for AC6 to increase cardiac responsiveness beneficially to β-AR stimulation is derived from transgenic mice with both constitutive and conditional cardiac-directed AC6 overexpression. These hearts displayed increased cAMP-generating capacity along with enhanced LV contractile function and relaxation in response to β-AR agonists. Basal performance and cAMP levels were not affected, and expression even at high levels (≤20-fold) for >20 months neither resulted in cardiac abnormalities nor affected longevity in mice. These results indicated a potentially favorable therapeutic window.
Advancement to murine HF models first unveiled AC6’s therapeutic potential; crossbreeding AC6 with Gαq transgenic mice that mimic clinical aspects of HF reversed decreased cAMP generation capacity, enhanced LV contractile performance as well as cardiac structural and electric remodeling, and increased survival. Correction of defects in β-AR–stimulated cAMP production and sarco(endo)plasmic reticulum (SR) Ca2+ uptake were of mechanistic importance for these functional improvements. In addition, AC6 seems to involve activating transcription factor 3 and pleckstrin homology domain leucine-rich protein phosphatase 2 for cAMP-independent actions. An AC6 mutant devoid of catalytic cAMP formation still decreased phospholamban (PLN) expression through activating transcription factor 3 activation and increased PLN phosphorylation at serin-16 through inhibition of the Akt phosphatase pleckstrin homology domain leucine-rich protein phosphatase 2, both relieving PLN-mediated attenuation of SERCA2 activity. AC6 apparently uses indirect SERCA2 activation for its therapeutic efficacy in HF. More mechanistic studies are clearly warranted to unearth underlying molecular mechanisms fully.
These results prompted testing of DNA-based strategies using AC6 expression in small and large animal HF models by means of cardiac-directed gene delivery. Intracoronary delivery of 2.5×1010 viral particles of AdAC6 to failing hearts of Gαqtransgenic mice showed improvement in LV systolic and diastolic contractile indices after 14 days. Subsequent testing of AdAC6 gene delivery in a porcine model showed >30% myocardial transduction rate when 1.4×1012 viral particles of AdAC6 were delivered via intracoronary infusion preceded by histamine application. Driven by a CMV promoter, LV AC6 protein content increased by ≈2-fold. Enhanced LV basal and β-AR–stimulated function persisted for ≥18 weeks including elevated cAMP production capacity. These results were recapitulated in a second large animal model of pacing-induced HF. Plasma brain natriuretic peptide concentrations were lower because of AC6 treatment but hearts showed no signs of reversed remodeling at that time. Interestingly, there is a previous report detailing antifibrotic effects by AC6 overexpression in fibroblasts with the use of an unbiased CMV promoter, so that the collateral effects of AC6 on cardiac fibroblast functions in failing hearts deserve future considerations.
AC6-DNA–based HF therapy was subsequently tested in human-relevant large animal models yielding promising results with respect to efficacy and safety. Despite the lack of quantitative data to describe PK/PD aspects of AdAC6 HF therapy, a current clinical phase I/II trial applies catheter-based intracoronary AdAC6 delivery to human failing hearts (Table). Cardiac inflammation previously reported for the use of Ad in myocardial gene delivery was not seen in AdAC6 large animal studies, but AdAC6 was applied at an ≈10- to 100-fold lower dosage per gram heart weight. This might explain persisting effects on cardiac performance for ≥18 weeks. Although failing hearts are generally more prone toward β-AR–mediated arrhythmias, there was no indication for this in preclinical studies using AdAC6. Results of the ongoing phase I/II trial will soon inform about safety and finding of efficient dosages.
Calcium (Ca)2+ Cycling as a Target System for HF Gene Therapy
Disturbed cardiomyocyte Ca2+ cycling emerged as hallmark of HF as failing human cardiomyocytes display characteristic alterations of their intracellular Ca2+ cycle. This includes decreased systolic Ca2+ levels followed by delayed diastolic Ca2+decay and elevated resting Ca2+ concentrations. Failing hearts subsequently generate a weak systolic contraction and prolonged diastolic relaxation with elevated stiffness impairing chamber filling. Pharmacological approaches such as levosimendan seeking to overcome systolic Ca2+ shortage by enhancing Ca2+sensitivity of thin filaments resulted in improved systolic force development but came at the cost of impaired diastolic Ca2+ dissociation that aggravated cardiac muscle relaxation and enhanced propensity toward malignant tachyarrhythmias.
Impaired activity of the SERCA2a/PLN complex and the EF-hand Ca2+ sensor protein S100A1 have emerged as key defects in abnormal cardiac Ca2+cycling. In view of their critical function, restoring both SERCA2a and S100A1 activity in failing hearts poses promising therapeutic DNA-based strategies to improve cardiac contractile performance (Figure 7).
Restoring SERCA2a Function
Failing human myocardium exhibits decreased SR Ca2+ uptake because of diminished SERCA2a expression. The major function of the SR Ca2+ pump in cardiomyocytes is diastolic SR Ca2+ reuptake and maintenance of SR Ca2+ load. Although the former is a crucial determinant of cardiac relaxation, the latter critically contributes to systolic performance. The important biological role in cardiac contraction regulation was shown specifically in heterozygous SERCA2a gene-ablated mice. An ≈2-fold decrease in cardiac SERCA2a expression significantly impaired cardiac contraction and relaxation. Vice versa, rodents with only a 1.6-fold cardiac-directed SERCA2a overexpression demonstrated increased global cardiac function without signs of long-term pathological consequences, and isolated cardiomyocytes yielded improved Ca2+ handling and enhanced SR Ca2+load.
Pathophysiological relevance of diminished cardiac SERCA2a levels was shown when heterozygous SERCA2a gene-ablated mice were subjected to experimental cardiac injury and hemodynamic stress. Mice with decreased SERCA2a expression both displayed increased MI size and worsened post-MI performance, and accelerated transition to HF in response to hemodynamic overload. Increased susceptibility of SERCA2a transgenic rats toward lethal ventricular arrhythmias after MI raised concerns about potential proarrhythmogenic side effects of the molecular inotrope, although surviving animals had improved cardiac performance.
The above-named studies provided a clear rationale to test targeted restoration of SERCA2a expression in diseased myocardium. In human failing cardiomyocytes, adenoviral-mediated SERCA2a-DNA (Ad-SERCa2a) delivery greatly improved cell shortening and Ca2+ handling. In rat hearts with chronic hemodynamic overload, antegrade coronary delivery of Ad-SERCA2a restored diminished SERCA2a protein to normal levels when a nonselective promoter element was used. LV systolic and diastolic performance was significantly enhanced and improvement in survival and energy potential of overloaded hearts was seen up to several weeks. Likewise, restoration of global SERCA2a protein expression in overloaded rat hearts was achieved using an AAV1-SERCA2a vector. Subsequently, LV function improved and oxygen cost of LV contractility within a 45-day follow-up was significantly reduced.
These beneficial effects prompted testing of long-term efficacy and safety in large animal models. Percutaneous catheter-based intracoronary delivery of 1×1012DNAse-resistant particles of AAV1-SERCA2a resulted in restoration of global LV SERCA2a protein levels in a pig model of experimental LV volume overload. Cardiac systolic and diastolic functional indices and structural remodeling were significantly improved within a 2-month follow-up. No myocardial toxicity and alterations in blood cell counts and clinical chemistry were reported, indicating safety of the viral-based HF therapy in a clinical-like setting. Potential extracardiac SERCA2a expression because of the use of a CMV promoter was not investigated. Prior concerns of an arrhythmogenic potential were weakened in Ad-SERCA2a–treated pigs showing reduced incidence of ventricular arrhythmias after ischemia/reperfusion injury.
In a sheep model of experimental LV volume overload, 5×1012 particles of AAV6-SERCA2a were intracoronarily delivered during balloon-mediated occlusion of cardiac venous outflow and prior adenosine application. AAV6-SERCA2a application restored global cardiac SERCA2a protein levels, resulting in better LV contractile indices and remodeling parameters within a 3-month follow-up. Myocardial transduction rate and potential extracardiac SERCA2a expression, as well as AAV6 biodistribution, were not reported. In addition, the usage of a cardiac-directed recirculating gene delivery methodology elicited dose-dependent increases in LV function and reversed remodeling in a pacing-induced sheep HF model when incremental amounts of AAV1-SERCA2a (1×1010, 1×1012, and 1×1013 DNAse-resistant particles) were applied. Neither clinical chemistry parameters nor blood cell counts were altered. Despite report of greatest therapeutic efficacy in the high-dosage group, the study did not investigate myocardial and extramyocardial distribution of SERCA2a protein to underline their results.
On the basis of sufficient data with respect to therapeutic efficacy, safety, and technical feasibility for cardiac-directed AAV-based gene delivery (Table), the CUPID trial has evaluated safety and efficacy of AAV1-SERCA2a in human chronic HF. Participants with advanced ischemic HF (New York Heart Association [NYHA] class III) enrolled in this trial received a single antegrade intracoronary infusion of AAV1-SERCA2a using a noncardioselective CMV promoter. CUPID phase I showed an acceptable safety profile, and previously published functional results and cumulative clinical event rates of phase II that randomized 39 patients with HF to 3 different dosages of intracoronary AAV1-SERCA2a (6×1011, 3×1012, and 1×1013DNase-resistant particles) versus placebo were encouraging. In light of limited statistical power, predefined multiple-efficacy domain analysis was applied showing improvement in the high-dosage AAV1-SERCA2a group versus control patients. Such domains take into consideration a series of parameters (symptoms, 6-minutes walk distance test, maximal oxygen consumption, brain natriuretic peptide levels, and echocardiographic measures). Greatly anticipated results of phase III will eventually allow further assessment of efficacy and affect morbidity. Other ongoing trials are currently recruiting patients to investigate effects of SERCA2a gene therapy on LV remodeling and on LV assist device therapy in patients with advanced HF.
Besides, targeted manipulation of SERCA2a expression is not the sole means to modulate SR Ca2+ uptake. Suppression of PLN was used as indirect approach to increase SERCA2a activity. Using RNA interference, an AAV9 vector generated stable production of a regulatory RNA sequence after being injected into the tail vein of rats. In this study, small hairpin RNA (shRNA) silenced cardiac PLN expression by ≈75%. More than 3 months, rats with transaortic banding–induced HF treated with AAV9-shPLN displayed improved cardiac function, reversed cardiac dilation and hypertrophy, and reduced cardiac fibrosis. Expression of shPLN was driven by a noncardioselective CMV promoter. Apparently, no adverse effects on microRNA and liver function were seen. Because a PLN null mutation is associated with cardiomyopathy in humans, forthcoming genetic strategies modulating SERCA2a activity by targeting PLN function are to be viewed with extreme caution. For further information on alternative strategies to target SERCA2 protein turnover and PLN phosphorylation, we refer to a previously published excellent review in this journal.
Improving S100A1 Activity
S100A1, an EF-hand Ca2+-sensor protein, is mostly expressed in cardiomyocytes. Failing human hearts show diminished expression of the EF-hand Ca2+-sensor raising interest in of S100A1’s role in cardiac muscle. A function in cardiac contractility regulation was first demonstrated by cardiac-targeted transgenic and viral-mediated S100A1 overexpression in mice and isolated cardiomyocytes, respectively, revealing inotropic and lusitropic effects. S100A1-mediated chronic gain-in-function is additive to β-AR stimulation but occurs cAMP independent. S100A1 controls a Ca2+-driven network in cardiomyocytes, which is key to the heart’s ability to contract and relax, to maintain cardiac mitochondrial energy homeostasis, and to withstand maladaptive growth and tachyarrhythmia. S100A1’s pleiotropic actions are mediated via direct binding and activity modulation of previously identified target proteins such as the ryanodine receptor 2, SERCA2a,cardiac titin, and numerous mitochondrial enzymes involved in cardiac energy homeostasis. S100A1/ryanodine receptor 2 binding improves excitation–contraction (ec) coupling gain and prevents diastolic SR Ca2+ leak. Interaction with the SERCA2a/PLN complex improves SR Ca2+ uptake and decreases diastolic Ca2+levels. Decreased stiffness and improved diastolic Ca2+ dissociation from cardiac myofilaments involves S100A1 binding to cardiac titin and attenuation of cardiac actin/titin interaction. S100A1 is also located in mitochondria improving ATP generation caused by F1-ATPase stimulation besides activity modulation of further mitochondrial enzymes.
In gene-ablated mice, cardiac S100A1 deletion worsened post-MI contractile performance basally and in response to β-AR stimulation, and post-MI survival was severely impaired. S100A1 overexpressing mice on the other hand, although having the same MI size, showed the opposite phenotype providing a strong rationale for therapeutic S100A1 restoration, potentially suitable for gene-based strategies. First proof-of-concept was demonstrated in an experimental rat HF model displaying diminished S100A1 protein levels comparable with human HF. Intracoronary delivery of a rAd5 carrying the human S100A1 cDNA under control of a CMV promoter was used and application of 1×1010 infectious viral particles was sufficient to normalize global cardiac S100A1 protein levels. Within 2 weeks, basal in vivo systolic and diastolic contractile performance of the heart was restored and β-AR responsiveness improved significantly, despite persistent signaling abnormalities. Approximately 40% to 50% myocardial transduction efficiency yielding inhomogeneous S100A1 expression distribution was sufficient to generate this therapeutic effect. Accordingly, intracellular Ca2+-transient amplitudes of AdS100A1-infected cardiomyocytes isolated from treated hearts were found to be either restored or increased above normal levels. S100A1 also decreased cytoplasmic Na+ levels that further contributed to improved mitochondrial ATP generation and reconstituted phosphocreatine/ATP ratio. Overall, this study unveiled a new therapeutic paradigm conveyed by S100A1 combining regeneration of contractile performance with reversal of maladaptive growth and energetic deficit.
First evidence for the same therapeutic efficacy of long-term cardiac-specific S100A1 restoration was demonstrated using intracoronary delivery of an AAV6, enabling sustained myocardial S100A1 expression with 40% to 50% transduction efficiency.29 A total of 2.5×1011 infectious viral particles of AAV6-S100A1 expressing the transgene under control of the cardiac-biased CARD-EF1 promoter restored overall cardiac S100A1 protein levels and prevented extracardiac S100A1 expression. Both systolic and diastolic contractile indices in S100A1-treated failing hearts were significantly improved within an 8-week follow-up period. Notably, the inotropic effect of S100A1 was preserved under concurrent β-AR blocker therapy. In contrast to β-AR blocker therapy alone, which only slowed down disease progression, AAV6-S100A1 therapy reversed contractile dysfunction. Both therapies were equally effective and when combined synergistic in protecting injured hearts from cardiac remodeling, but only S100A1-treated hearts showed improved β-AR responsiveness.
Using an experimental porcine post-MI HF model, cardiac-directed delivery of 1.5×1013 AAV9-S100A1 particles 2 weeks after MI via the anterior coronary vein resulted in wide-spread S100A1 expression. The use of the cardiac-biased CMVmini-enh.-MLCV2 expression cassette restricted S100A1 expression to the heart ranging from normal to 5- to 6-fold overexpression in targeted areas. Restoring S100A1 in failing pig hearts yielded significant long-term improvement both of systolic and diastolic cardiac function after 3 months. Functional regeneration was mirrored by increased Ca2+ transient amplitudes in isolated cardiomyocytes from S100A1-treated hearts. AAV9-S100A1 treatment, furthermore, reversed myocardial remodeling and normalized the decreased phosphocreatine/ATP ratio indicative of improved cardiac energy homeostasis. Comprehensive biomarker assessment showed unchanged clinical chemistry, blood count, and coagulation markers in addition to normal peripheral organ histology and indicated safety of AAV9-S100A1 therapy. In support of its efficacy in human myocardium, effects of S100A1 restoration in human failing cardiomyocytes by Ad5-mediated human S100A1 cDNA delivery corroborated the previous findings entirely.
Systematic analysis of S100A1’s therapeutic potency and mechanisms yielded strong evidence for its efficacy and safety in human-relevant HF models (Table). Notably, a moderate S100A1 gene dosage is already sufficient for normalized expression yielding significant therapeutic effects and the inotropic therapy is not associated with increases in heart rate or arrhythmias. S100A1’s therapeutic window tested to date in failing cardiomyocytes and hearts ranges from expression restoration to ≤10-fold overexpression without occurrence of adverse effects. Refinement of cardiac retrograde delivery technology and development of companion diagnostics for AAV9-S100A1 therapy is expected to facilitate translation to phase I/II trials.
For the discussion of DNA/RNA-based strategies aimed at manipulation of (1) stem cell homing and survival for cardiac regeneration, (2) angiogenesis, and (3) oxidative stress and resistance to cell death, we refer to already published excellent reviews.
Source ahajournals.org
DUC TIN Surgical Clinic
Tin tức liên quan
Performance diagnostique de l’interféron gamma dans l’identification de l’origine tuberculeuse des pleurésies exsudatives
A Mixed Phenotype of Airway Wall Thickening and Emphysema Is Associated with Dyspnea and Hospitalization for Chronic Obstructive Pulmonary Disease.
Radiological Approach to Asthma and COPD-The Role of Computed Tomography.

Significant annual cost savings found with UrgoStart in UK and Germany
Thrombolex announces 510(k) clearance of Bashir catheter systems for thromboembolic disorders
Phone: (028) 3981 2678
Mobile: 0903 839 878 - 0909 384 389