





Background
Tetralogy of Fallot (TOF) is one of the most common congenital heart disorders (CHDs). This condition is classified as a cyanotic heart disorder, because tetralogy of Fallot results in an inadequate flow of blood to the lungs for oxygenation (right-to-left shunt) (see the following image). Patients with tetralogy of Fallot initially present with cyanosis shortly after birth, thereby attracting early medical attention.
Tetralogy of Fallot (TOF) is one of the most common congenital heart disorders (CHDs). This condition is classified as a cyanotic heart disorder, because tetralogy of Fallot results in an inadequate flow of blood to the lungs for oxygenation (right-to-left shunt) (see the following image). Patients with tetralogy of Fallot initially present with cyanosis shortly after birth, thereby attracting early medical attention.
Typical features
The 4 features typical of tetralogy of Fallot include right ventricular (RV) outflow tract obstruction (RVOTO) (infundibular stenosis), ventricular septal defect (VSD), aorta dextroposition, and right ventricular hypertrophy. Occasionally, a few children also have an atrial septal defect (ASD), which makes up the pentad of Fallot. The basic pathology of tetralogy is due to the underdevelopment of the right ventricular infundibulum, which results in an anterior-leftward malalignment of the infundibular septum. This malalignment determines the degree of RVOTO.
The clinical features of tetralogy of Fallot are generally typical, and a preliminary clinical diagnosis can almost always be made. Because most infants with this disorder require surgery, it is fortunate that the availability of cardiopulmonary bypass (CPB), cardioplegia, and surgical techniques is now well established. Most surgical series report excellent clinical results with low morbidity and mortality rates
Historical information
Louis Arthur Fallot, after whom the name tetralogy of Fallot is derived, was not the first person to recognize the condition. Stensen first described it in 1672; however, it was Fallot who first accurately described the clinical and complete pathologic features of the defects.
Although the disorder was clinically diagnosed much earlier, no treatment was available until the 1940s. Cardiologist Helen Taussig recognized that cyanosis progressed and inevitably led to death in infants with tetralogy of Fallot. She postulated that the cyanosis was due to inadequate pulmonary blood flow. Her collaboration with Alfred Blalock led to the first type of palliation for these infants. In 1944, Blalock operated on an infant with tetralogy of Fallot and created the first Blalock-Taussig shunt between the subclavian artery and the pulmonary artery (see the image below).
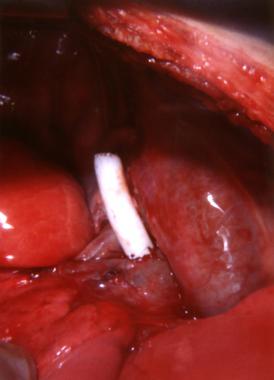
This image shows completed blocking with a Taussig shunt
The pioneering Blalock-Taussig shunt surgical technique opened a new era in neonatal cardiac surgery. Development of the Potts shunt (from the descending aorta to the left pulmonary artery), the Glenn shunt (from the superior vena cava to the right pulmonary artery), and the Waterston shunt (from the ascending aorta to the right pulmonary artery) followed.
Scott performed the first open correction in 1954. Less than half a year later, Lillehei performed the first successful open repair for tetralogy of Fallot using controlled cross-circulation, with another patient serving as oxygenator and blood reservoir. The following year, with the advent of cardiopulmonary bypass by Gibbons, another historic era of cardiac surgery was established. Since then, numerous advances in surgical technique and myocardial preservation have evolved in the treatment of tetralogy of Fallot.
Anatomy
Patients with tetralogy of Fallot (TOF) can present with a broad range of anatomic deformities. Fallot initially described 4 major defects consisting of : (1) pulmonary artery stenosis, (2) ventricular septal defect (VSD), (3) deviation of the aortic origin to the right, and (4) right ventricular hypertrophy (RV).
In the present day, however, the most important features of tetralogy of Fallot are recognized as (1) the right ventricular (RV) outflow tract obstruction (RVOTO), which is nearly always infundibular and/or valvular, and (2) an unrestricted VSD associated with malalignment of the conal septum (see the following image).
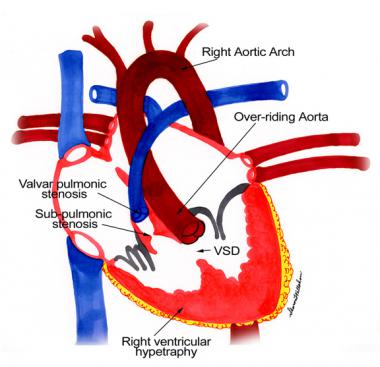
Anatomic findings in tetralogy of Fallot.
Right ventricle outflow tract obstruction
Clinically, most patients with tetralogy of Fallot have an increased resistance to right ventricle emptying because of the pulmonary outflow tract obstruction. The anterior displacement and rotation of the infundibular septum causes RV obstruction of variable degree and location. The obstruction may be adjacent to the pulmonary valve, causing additional obstruction.
Pulmonary arteries
The pulmonary arteries can vary in size and distribution, and they may be atretic or hypoplastic. Rarely, the left pulmonary artery is absent. In some individuals, a varying degree of stenosis of the peripheral pulmonary arteries occurs, which further restricts pulmonary blood flow.
Pulmonary atresia results in no communication between the RV and the main pulmonary artery; in this case, pulmonary blood flow is maintained by either the ductus arteriosus or collateral circulation from the bronchial vessels. With minimal RVOTO, pulmonary vascular disease may develop secondary to excessive pulmonary blood flow from the large left-to-right shunt or large aortopulmonary collaterals.
In up to 75% of children with tetralogy of Fallot, some degree of pulmonary valve stenosis may occur. Stenosis is usually due to leaflet tethering rather than commissural fusion. The pulmonary annulus is narrowed in virtually every case.
Aorta
True dextroposition and abnormal rotation of the aortic root result in aortic overriding (ie, an aorta that, to varying degrees, originates from the RV). In some cases, more than 50% of the aorta may thus originate from the RV. A right aortic arch may occur, which may lead to an abnormal origin of the arch vessels.
Associated anomalies
Associated defects are also common. The coexistence of an atrial septal defect (ASD) occurs often enough to prompt its inclusion in a so-called pentalogy of Fallot. Other possible defects include patent ductus arteriosus (PDA), atrioventricular septal defects (AVSD), muscular VSD, anomalous pulmonary venous return, anomalous coronary arteries, absent pulmonary valve, aorticopulmonary window, and aortic incompetence.
The coronary anatomy may also be abnormal. Among these abnormalities is the origin of the left anterior descending (LAD) coronary artery from the proximal right coronary artery (PRCA), which crosses the RV outflow at variable distances from the pulmonary valve annulus. The anomalous LAD coronary artery is observed in 9% of cases of tetralogy of Fallot, and this abnormality makes placement of a patch across the pulmonary annulus risky, possibly requiring an external conduit. During the VSD repair, the anomalous LAD coronary artery is prone to injury. Occasionally, all coronary arteries arise from a single left main coronary ostium.
Etiology and Pathophysiology
The cause(s) of most congenital heart diseases (CHDs) are unknown, although genetic studies suggest a multifactorial etiology. A study from Portugal reported that methylene tetrahydrofolate reductase (MTHFR) gene polymorphism can be considered a susceptibility gene for tetralogy of Fallot. A more recent study has reported that VEGF genetic polymorphisms, -2578C>A and -634C>G, may be associated with an increased risk for tetralogy of Fallot, whereas the risk is potentially reduced with 936C>T polymorphism.
Prenatal factors associated with a higher incidence of tetralogy of Fallot (TOF) include maternal rubella (or other viral illnesses) during pregnancy, poor prenatal nutrition, maternal alcohol use, maternal age older than 40 years, maternal phenylketonuria (PKU) birth defects, and diabetes. Children with Down syndrome also have a higher incidence of tetralogy of Fallot, as do infants with fetal hydantoin syndrome or fetal carbamazepine syndrome.
As one of the conotruncal malformations, tetralogy of Fallot can be associated with a spectrum of lesions known as CATCH 22 (cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia). Cytogenetic analysis may demonstrate deletions of a segment of chromosome band 22q11 (DiGeorge critical region). Ablation of cells of the neural crest has been shown to reproduce conotruncal malformations.
These abnormalities are associated with the DiGeorge syndrome and branchial arch abnormalities.
The hemodynamics of tetralogy of Fallot depend on the degree of right ventricular (RV) outflow tract obstruction (RVOTO). The ventricular septal defect (VSD) is usually nonrestrictive, and the RV and left ventricular (LV) pressures are equalized. If the obstruction is severe, the intracardiac shunt is from right to left, and pulmonary blood flow may be markedly diminished. In this instance, blood flow may depend on the patent ductus arteriosus (PDA) or bronchial collaterals.
Prognosis
Early surgery is not indicated for all infants with tetralogy of Fallot (TOF), although, without surgery, the natural progression of the disorder indicates a poor prognosis. The progression of the disorder depends on the severity of right ventricular (RV) outflow tract obstruction (RVOTO).
In the present era of cardiac surgery, children with simple forms of tetralogy of Fallot enjoy good long-term survival with an excellent quality of life. Late outcome data suggest that most survivors are in New York Heart Association (NYHA) classification I, although maximal exercise capability is reduced in some
Sudden death from ventricular arrhythmias has been reported in 1-5% of patients at a later stage in life, and the cause remains unknown. It has been suspected that ventricular dysfunction may be the cause. One study found left ventricular longitudinal dysfunction to be associated with a greater risk of developing life-threatening arrhythmias. Continued cardiac monitoring into adult life is necessary. For some time, it has been suspected that certain children may have inherited a predispostion to developing long QT syndrome. A 2012 study by Chiu confirmed this suspicion.
If left untreated, patients with tetralogy of Fallot face additional risks that include paradoxical emboli leading to stroke, pulmonary embolus, and subacute bacterial endocarditis. It is well known that children with congenital heart disease are prone to stroke. In most of these children the causes of stroke have been related to thromboemboli, prolonged hypotension/anoxix and polycythemia. What is often forgotten is that residual shunts or a patent foramen ovale are also known causes of strokes. The investigation of strokes in these children usually begins with a CT scan of the brain followed by an ECHO.
Without surgery, mortality rates gradually increase, ranging from 30% at age 2 years to 50% by age 6 years. The mortality rate is highest in the first year and then remains constant until the second decade. No more than 20% of patients can be expected to reach the age of 10 years, and fewer than 5-10% of patients are alive by the end of their second decade.
Most individuals who survive to age 30 years develop congestive heart failure (CHF), although individuals whose shunts produce minimal hemodynamic compromise have been noted, albeit rarely, and these individuals achieve a normal life span. However, cases of survival of patients into their 80s have been reported. Due to advanced surgical techniques, a 40% reduction in deaths associated with tetralogy of Fallot was noted from 1979 to 2005.
As might be expected, individuals with tetralogy of Fallot and pulmonary atresia have the worst prognoses, and only 50% survive to age 1 year and 8% to age 10 years.
Source emedicine.com
Duc Tin Surgical Clinic
Tin tức liên quan
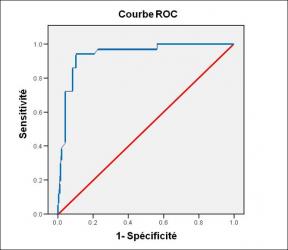
Performance diagnostique de l’interféron gamma dans l’identification de l’origine tuberculeuse des pleurésies exsudatives
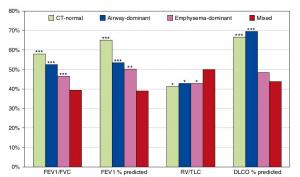
A Mixed Phenotype of Airway Wall Thickening and Emphysema Is Associated with Dyspnea and Hospitalization for Chronic Obstructive Pulmonary Disease.
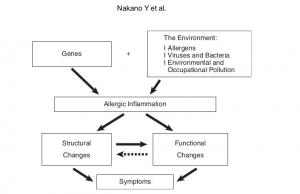
Radiological Approach to Asthma and COPD-The Role of Computed Tomography.

Significant annual cost savings found with UrgoStart in UK and Germany
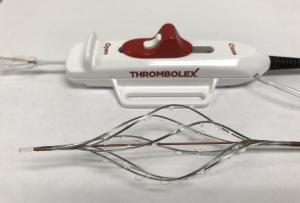
Thrombolex announces 510(k) clearance of Bashir catheter systems for thromboembolic disorders
Phone: (028) 3981 2678
Mobile: 0903 839 878 - 0909 384 389